






Avaliação clínica de lesões de nervos periféricos: "
 Por Bruno Melo Fernandes
Por Bruno Melo Fernandes
Resumo
EM sua modalidade de polineuropatia, afetan vários nervos de forma simultânea, con sinais simétricos distais e participação de pares cranianos ou não. São anormalidades neurológicas frequentes e sua gravidade varia desde anormalidades sensoriais leves até transtornos paralíticos graves, como a Síndrome de Guillain Barré (CASTAÑEDA-FERNANDÉZ et al., 2003).
A avaliação da neuropatia diabética, por sua vez, é um desafio. Esta complicação é geralmente avaliada de maneira subjetiva (sintomas inespecíficos) e não padronizada. Em alguns casos, a presença de alterações nos reflexos e na sensibilidade pode facilitar o diagnóstico. Estes sinais, contudo, não estão presentes em todos os pacientes. O método mais comum de avaliação da avaliação da neuropatia diabética é o uso do teste do monofilamento, exame que avalia a sensibilidade tátil nos pés e pododáctilos dos pacientes (MEULENBELT et al., 2009). Os outros métodos recomendados são complexos e de mais difícil aplicação na prática clínica.
É importante perceber que a avaliação sensitiva à beira do leito, isto é, o exame físico, é menos sensível e confiável que a percepção própria do paciente, esta denotada pela anamnese. Em muitos casos, é mais fácil definir a anatomia exata de uma área de sensibilidade alterada pedindo-se ao paciente para desenhar a região afetada sobre o corpo. Esses achados são mais confiáveis que os do exame físico, caso estes sejam diferentes.
Em quadros mais tardios, os sintomas sensitivos podem se juntar a sintomas motores, em especial, a paresia, acompanhada ou não de atrofia. Deve-se atentar para o fato de que nem todos os músculos distais a uma lesão nervosa são afetados, pois não necessariamente todas as fibras do nervo lesionado estão disfuncionantes.
A avaliação clínica de uma lesão de nervo periférico deve então priorizar a busca por sintomas referidos pelo paciente que possam demonstrar déficits clínicos específicos à distribuição de nervos ou segmentos nervosos. Tais suspeitas podem ou não serem confirmadas através de estudo eletroneuromiográfico, a depender da dúvida quanto ao diagnóstico. Além disso, deve-se buscar uma etiologia para o processo, como a história de trauma ou um hábito de vida (como tocar violino ou jogar boliche), ou seja, atividades ligadas a determinadas neuropatias específicas.
É importante salientar que quanto à etiologia traumática, estima-se em 10% a 20% a prevalência de lesões do sistema nervoso periférico envolvem o plexo braquial. Os mecanismos de tração/estiramento foram os principais responsáveis por lesão do plaxo braquial. Os traumas de plexo braquial provocam disfunção tanto por lesão neurológica quanto por desenvolvimento de dor no membro afetado. A dor, nesses casos, pode ocorrer por três mecanismos: dor neuropática em lesões pós-ganglionares; dor devido a avulsão radicular; ou dor simpático-mediada (FLORES, 2006).
O diagnóstico precoce é imperativo para uma abordagem terapêutica eficaz, que passam por mudanças em hábitos de vida, como posturas ou atividades, uso de suportes ou coletes específicos, sessões de fisioterapia e, em alguns casos, abordagens cirúrgicas específicas.
Referências
CASTAÑEDA-FERNANDÉZ, J. A; CORRA-GARCÍA, J. Neuropatías periféricas. Medisan, 7 (4), s. p., 2003. Disponível em: http://bvs.sld.cu/revistas/san/vol7_4_03/san07403.htm. Acesso em 22 mar. 2011.
FLORES, L. P. Estudo epidemiológico das lesões traumáticas de plexo braquial em adultos. Arq Neuropsiquiatr, 64(1):88-94, 2006.
KASPARYAN G.N., RUSSEL J. A. Aspectos Gerais das Momoneuropatias. In: HOYDEN JONES JR, H. et al. (Ed.) Neurologia de Netter. Porto Alegre: Artmed, 2007. Cap 83.
MARGLES S.W., RUSSEL J. A. Mononeuropatias que apresentam sintomas nos membros superiores. In: HOYDEN JONES JR, H. et al. (Ed.) Neurologia de Netter. Porto Alegre: Artmed, 2007. Cap 83.
MATTAR JÚNIOR, R.; AZZE, R. J. Lesões dos nervos periféricos. Atualização em Traumatologia do Aparelho Locomotor. 2008. Disponível em: http://www.ronaldoazze.com.br/fasciculo/fasciculo3.PDF. Acesso em: 22 mar. 2011.
MEULENBELT, M. et al. Determinants of skin problems of the stump in lower-limb amputees. Arch Phys Med Rehabil, 90 (1):74-81, 2009.
fonte da postagem:http://semiologiamedica.blogspot.com/2011/03/avaliacao-clinica-de-lesoes-de-nervos.html "
"
 Por Bruno Melo Fernandes
Por Bruno Melo FernandesEstudante de Graduação em Medicina da UFPB
Resumo
As lesões de nervos periféricos estão entre as desordens neurológicas mais comuns. De etiologias traumáticas e não-traumáticas, as neuropatias periféricas podem se manifestar clinicamente com uma significativa variedade de sintomas: sensação de formigamento, dormência, queimação, dor, hiperestesia, redução da força muscular, atrofias, hipotensão postural, disfunção erétil, anidrose, incontinência esfincteriana, entre otros sintomas e sinais. É importante que o médico conheça as funções, e os mecanismos de lesão, dos principais nervos periféricos para que, assim, possa diagnosticar neuropatias a partir de achados puramente clínicos.
Palavras-chave: Nervos Periféricos. Neuropatia Periférica. Técnicas de Diagnóstico Neurológico.
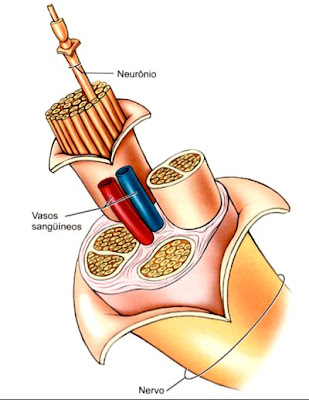
O sistema nervoso periférico é composto por todos os nervos que estão fora do sistema nervoso central (cérebro e medula espinhal): raízes, gânglios, plexos, fibras nervosas. Estes elementos neurais são extensões do sistema nervoso central responsáveis pela integração das atividades nervosas não axiais, tanto sensitivas quanto motoras. Portanto, esses nervos são uma linha de condução que leva informações sensoriais da pele, músculos e outros órgãos ao sistema nervoso central (SNC) e informações motoras do SNC para os músculos somáticos e órgãos efetores controlados pelo sistema nervoso autônomo.
As lesões de nervos periféricos, traumáticas ou não, acarretam perdas parciais ou totais de numerosas funções orgânicas, devido à parada da transmissão de impulsos nervosos e à desorganização funcional dos tecidos acometidos (MATTAR JÚNIOR; AZZE, 2008).
Em relação à clínica médica, as lesões de nervos periféricos formam um quadro de afecções extremamente diverso, com manifestações clínicas e etiologias múltiplas, sendo muito comumente encontradas na prática médica. Em geral, dividem-se as lesões de nervos periféricos em traumáticas e não-traumáticas, diferença esta facilmente identificada na avaliação clínica dos pacientes (KARPARIAN; RUSSEL, 2007).
As neuropatias periféricas podem se manifestar clinicamente com uma significativa variedade de sintomas: sensação de formigamento, dormência, queimação, dor, hiperestesia, redução da força muscular, atrofias, hipotensão postural, disfunção erétil, anidrose, incontinência esfincteriana, entre otros sintomas e sinais.
O primeiro passo na abordagem clínica de lesões de nervos periféricos é a anamnese. O médico deve atentar para os sintomas referidos pelo paciente, seu tempo e curso de evolução e, principalmente, tentar encontrar algum evento desencadeador do problema, como um hábito de vida, um trauma, uma doença hereditária, ou um antecedente pessoal patológico, entre outras causas.
Em geral, as lesões de nervos periféricos acarretam distúrbios focais das funções motora, sensitiva e autonômica e, às vezes, dor. O quadro clínico de uma neuropatia varia de acordo com os tipos de fibras danificados no nervo envolvido e com a fisiopatologia da lesão. As lesões podem ser agudas ou de apresentação insidiosa e ainda podem acarretar sintomas intermitentes, como no caso de isquemia nervosa transitória de origem mecânica, ou constantes, como no caso de ruptura de axônios (Ibid).
Acredita-se que as fibras sensitivas são mais suscetíveis a lesões que as motoras localizadas ao lado delas. Logo, é comum que os primeiros sintomas de uma mononeuropatia sejam sensitivos. Os pacientes podem se queixar de perda de sensibilidade ou anestesia, “dormência”, parestesias, sensação de inchaço, ou sensações anormais tipo queimação, hipersensibilidade e prurido (MARGLESS; RUSSEL, 2007).
O sistema nervoso periférico é composto por todos os nervos que estão fora do sistema nervoso central (cérebro e medula espinhal): raízes, gânglios, plexos, fibras nervosas. Estes elementos neurais são extensões do sistema nervoso central responsáveis pela integração das atividades nervosas não axiais, tanto sensitivas quanto motoras. Portanto, esses nervos são uma linha de condução que leva informações sensoriais da pele, músculos e outros órgãos ao sistema nervoso central (SNC) e informações motoras do SNC para os músculos somáticos e órgãos efetores controlados pelo sistema nervoso autônomo.
As lesões de nervos periféricos, traumáticas ou não, acarretam perdas parciais ou totais de numerosas funções orgânicas, devido à parada da transmissão de impulsos nervosos e à desorganização funcional dos tecidos acometidos (MATTAR JÚNIOR; AZZE, 2008).
Em relação à clínica médica, as lesões de nervos periféricos formam um quadro de afecções extremamente diverso, com manifestações clínicas e etiologias múltiplas, sendo muito comumente encontradas na prática médica. Em geral, dividem-se as lesões de nervos periféricos em traumáticas e não-traumáticas, diferença esta facilmente identificada na avaliação clínica dos pacientes (KARPARIAN; RUSSEL, 2007).
As neuropatias periféricas podem se manifestar clinicamente com uma significativa variedade de sintomas: sensação de formigamento, dormência, queimação, dor, hiperestesia, redução da força muscular, atrofias, hipotensão postural, disfunção erétil, anidrose, incontinência esfincteriana, entre otros sintomas e sinais.
O primeiro passo na abordagem clínica de lesões de nervos periféricos é a anamnese. O médico deve atentar para os sintomas referidos pelo paciente, seu tempo e curso de evolução e, principalmente, tentar encontrar algum evento desencadeador do problema, como um hábito de vida, um trauma, uma doença hereditária, ou um antecedente pessoal patológico, entre outras causas.
Em geral, as lesões de nervos periféricos acarretam distúrbios focais das funções motora, sensitiva e autonômica e, às vezes, dor. O quadro clínico de uma neuropatia varia de acordo com os tipos de fibras danificados no nervo envolvido e com a fisiopatologia da lesão. As lesões podem ser agudas ou de apresentação insidiosa e ainda podem acarretar sintomas intermitentes, como no caso de isquemia nervosa transitória de origem mecânica, ou constantes, como no caso de ruptura de axônios (Ibid).
Acredita-se que as fibras sensitivas são mais suscetíveis a lesões que as motoras localizadas ao lado delas. Logo, é comum que os primeiros sintomas de uma mononeuropatia sejam sensitivos. Os pacientes podem se queixar de perda de sensibilidade ou anestesia, “dormência”, parestesias, sensação de inchaço, ou sensações anormais tipo queimação, hipersensibilidade e prurido (MARGLESS; RUSSEL, 2007).
EM sua modalidade de polineuropatia, afetan vários nervos de forma simultânea, con sinais simétricos distais e participação de pares cranianos ou não. São anormalidades neurológicas frequentes e sua gravidade varia desde anormalidades sensoriais leves até transtornos paralíticos graves, como a Síndrome de Guillain Barré (CASTAÑEDA-FERNANDÉZ et al., 2003).
A avaliação da neuropatia diabética, por sua vez, é um desafio. Esta complicação é geralmente avaliada de maneira subjetiva (sintomas inespecíficos) e não padronizada. Em alguns casos, a presença de alterações nos reflexos e na sensibilidade pode facilitar o diagnóstico. Estes sinais, contudo, não estão presentes em todos os pacientes. O método mais comum de avaliação da avaliação da neuropatia diabética é o uso do teste do monofilamento, exame que avalia a sensibilidade tátil nos pés e pododáctilos dos pacientes (MEULENBELT et al., 2009). Os outros métodos recomendados são complexos e de mais difícil aplicação na prática clínica.
É importante perceber que a avaliação sensitiva à beira do leito, isto é, o exame físico, é menos sensível e confiável que a percepção própria do paciente, esta denotada pela anamnese. Em muitos casos, é mais fácil definir a anatomia exata de uma área de sensibilidade alterada pedindo-se ao paciente para desenhar a região afetada sobre o corpo. Esses achados são mais confiáveis que os do exame físico, caso estes sejam diferentes.
Em quadros mais tardios, os sintomas sensitivos podem se juntar a sintomas motores, em especial, a paresia, acompanhada ou não de atrofia. Deve-se atentar para o fato de que nem todos os músculos distais a uma lesão nervosa são afetados, pois não necessariamente todas as fibras do nervo lesionado estão disfuncionantes.
A avaliação clínica de uma lesão de nervo periférico deve então priorizar a busca por sintomas referidos pelo paciente que possam demonstrar déficits clínicos específicos à distribuição de nervos ou segmentos nervosos. Tais suspeitas podem ou não serem confirmadas através de estudo eletroneuromiográfico, a depender da dúvida quanto ao diagnóstico. Além disso, deve-se buscar uma etiologia para o processo, como a história de trauma ou um hábito de vida (como tocar violino ou jogar boliche), ou seja, atividades ligadas a determinadas neuropatias específicas.
É importante salientar que quanto à etiologia traumática, estima-se em 10% a 20% a prevalência de lesões do sistema nervoso periférico envolvem o plexo braquial. Os mecanismos de tração/estiramento foram os principais responsáveis por lesão do plaxo braquial. Os traumas de plexo braquial provocam disfunção tanto por lesão neurológica quanto por desenvolvimento de dor no membro afetado. A dor, nesses casos, pode ocorrer por três mecanismos: dor neuropática em lesões pós-ganglionares; dor devido a avulsão radicular; ou dor simpático-mediada (FLORES, 2006).
O diagnóstico precoce é imperativo para uma abordagem terapêutica eficaz, que passam por mudanças em hábitos de vida, como posturas ou atividades, uso de suportes ou coletes específicos, sessões de fisioterapia e, em alguns casos, abordagens cirúrgicas específicas.
Referências
CASTAÑEDA-FERNANDÉZ, J. A; CORRA-GARCÍA, J. Neuropatías periféricas. Medisan, 7 (4), s. p., 2003. Disponível em: http://bvs.sld.cu/revistas/san/vol7_4_03/san07403.htm. Acesso em 22 mar. 2011.
FLORES, L. P. Estudo epidemiológico das lesões traumáticas de plexo braquial em adultos. Arq Neuropsiquiatr, 64(1):88-94, 2006.
KASPARYAN G.N., RUSSEL J. A. Aspectos Gerais das Momoneuropatias. In: HOYDEN JONES JR, H. et al. (Ed.) Neurologia de Netter. Porto Alegre: Artmed, 2007. Cap 83.
MARGLES S.W., RUSSEL J. A. Mononeuropatias que apresentam sintomas nos membros superiores. In: HOYDEN JONES JR, H. et al. (Ed.) Neurologia de Netter. Porto Alegre: Artmed, 2007. Cap 83.
MATTAR JÚNIOR, R.; AZZE, R. J. Lesões dos nervos periféricos. Atualização em Traumatologia do Aparelho Locomotor. 2008. Disponível em: http://www.ronaldoazze.com.br/fasciculo/fasciculo3.PDF. Acesso em: 22 mar. 2011.
MEULENBELT, M. et al. Determinants of skin problems of the stump in lower-limb amputees. Arch Phys Med Rehabil, 90 (1):74-81, 2009.
fonte da postagem:http://semiologiamedica.blogspot.com/2011/03/avaliacao-clinica-de-lesoes-de-nervos.html








